

![]()

Built on top of SpatialData, Sopa enables processing and analyses of image-based spatial-omics using a standard data structure and output. We currently support the following technologies: Xenium, MERSCOPE, CosMX, PhenoCycler, MACSima, Hyperion. Sopa was designed for generability and low memory consumption on large images (scales to 1TB+ images).
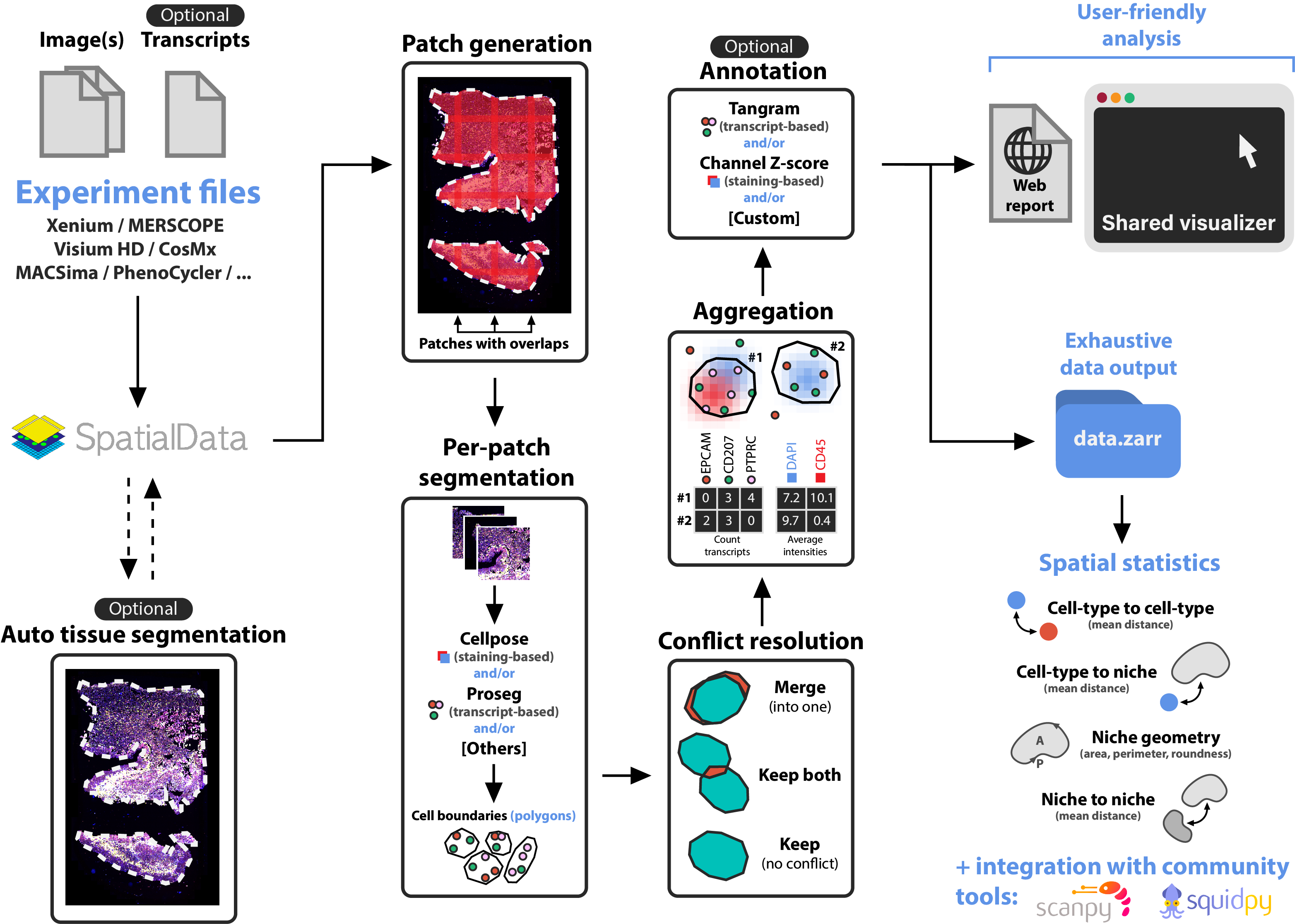
The pipeline outputs contain: (i) Xenium Explorer files for interactive visualization, (ii) an HTML report for quick quality controls, and (iii) a SpatialData .zarr directory for further analyses.
The easiest way to start with sopa is to check our documentation. It contains installation explanations, CLI/API details, and tutorials.
The following illustration describes the main steps of sopa:
Sopa can be installed via PyPI on all operating systems. Make sure you have an environment with python==3.10 (more versions will be soon supported), and run the following command:
pip install sopa
To install extras (for example, if you want to use snakemake/cellpose/baysor/tangram), please run:
pip install 'sopa[snakemake,cellpose,baysor,tangram]'
Important: even though pip install 'sopa[baysor]' will install some dependencies related to baysor, you still have to install the baysor command line (see the official repository) if you want to use it.
You can clone the repository and run one of these command lines at the root of sopa:
pip install -e . # dev mode installation
poetry install # poetry installation
Sopa comes in three different flavours, each corresponding to a different use case:
Snakemake pipeline: choose a config, and run our pipeline on your spatial data in a couple of minutesCLI: use our command-line-interface for prototyping quickly your own pipelineAPI: use directlysopaas a Python package for complete flexibility and customization
Clone our repository, choose a config here (or create your own), and execute our pipeline locally or on a high-performance cluster:
git clone https://github.com/gustaveroussy/sopa.git
cd sopa/workflow
snakemake --configfile=/path/to/yaml_config --config data_path=/path/to/data_directory --cores 1 --use-condaFor more details on snakemake configuration and how to properly setup your environments, please refer to the documentation.
Below are examples of commands that can be run with the sopa CLI:
> sopa --help # show command names and arguments
> sopa read merscope_directory --technology merscope # read some data
> sopa patchify image merscope_directory.zarr # make patches for low-memory segmentation
> sopa segmentation cellpose merscope_directory.zarr --diameter 60 --channels DAPI # segmentation
> sopa resolve cellpose merscope_directory.zarr # resolve segmentation conflicts at boundaries
> sopa aggregate merscope_directory.zarr --average-intensities # transcripts/channels aggregation
> sopa explorer write merscope_directory.zarr # convert for interactive vizualisationFor a complete description of the CLI, please refer to the documentation.
import sopa
# use the 'sopa' python packageFor a complete API description, please refer to the documentation.
Our article is not published yet. In the meantime, you can cite our preprint:
@article {Blampey2023.12.22.571863,
author = {Quentin Blampey & Kevin Mulder et al.},
title = {Sopa: a technology-invariant pipeline for analyses of image-based spatial-omics},
elocation-id = {2023.12.22.571863},
year = {2023},
doi = {10.1101/2023.12.22.571863},
publisher = {Cold Spring Harbor Laboratory},
URL = {https://www.biorxiv.org/content/early/2023/12/23/2023.12.22.571863},
eprint = {https://www.biorxiv.org/content/early/2023/12/23/2023.12.22.571863.full.pdf},
journal = {bioRxiv}
}